
Huntington’s disease affects the brain and is always inherited. The only treatments for HD manage symptoms, some of them prescribed off-label, borrowed from other conditions. A treatment that addresses the underlying cause of the disease, which delays onset or slows progression, has been elusive for decades.
A disease like no other
HD is one of 40 “expanding repeat” diseases. A tiny part of a gene repeats many times, resulting in an encoded protein burdened with extra amino acids that interfere with its folding, rendering the protein sticky. In HD, mutant huntingtin protein gums up neurons in the brain’s striatum, blocking signals essential to control movements and to think. Behavior changes too – anger and aggression may soar, as irritability, loss of impulse control, and confusion reign. The white matter part of the brain – the axons of those neurons – shrinks.
In HD, a triplet of DNA bases – CAG – in the huntingtin gene repeats more than the normal 35 times. The more extra CAG copies the younger symptom onset, the more severe the uncontrollable movements, and the faster the physical and mental deterioration.
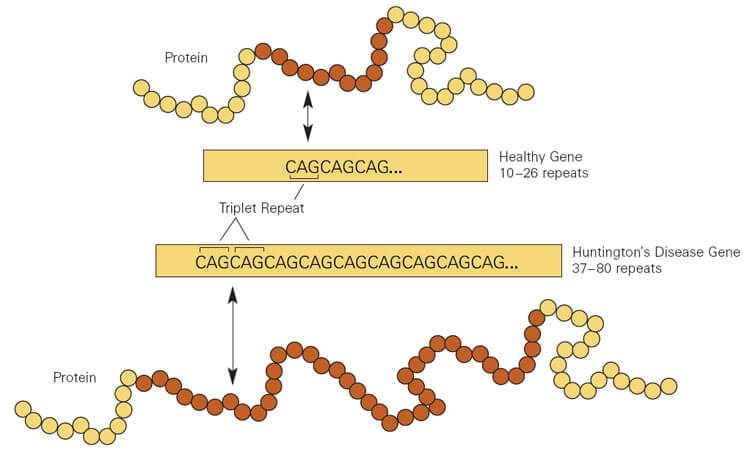
HD is inherited as an autosomal dominant: each child of an affected individual stands a 50:50 chance of having inherited the disease. An affected person has both a normal and mutant copy of the gene.
Because symptom onset is typically in adulthood, before predictive testing became available in the mid 1990s, people would have children before they knew they were destined to develop the disease. DNA Science covered the history of HD, and what it’s like to lose family to it, here.
Considering this clinical picture of devastation against a backdrop of a dearth of treatments for a rare disease that might not get much attention in the midst of a pandemic, setbacks in clinical trials are especially hard to take.
Drug trials halted
On March 22, Roche ceased treating new patients in its phase 3 clinical trial of tominersen, following recommendation from an independent data monitoring committee, part of the normal trajectory towards drug approval. Tominersen is a small molecule designed to glom onto the extra parts of the huntingtin gene, blocking it so that a near-normal sized gene is available for transcription into RNA and then translation into huntingtin protein. DNA Science covered the launch of the phase 1 clinical trial in 2015, from Isis Pharmaceuticals, which rebranded to Ionis with the rise of the terrorist group. Roche partnered with Ionis in December 2017 to further develop the drug.

No one was harmed from tominersen, but so far the data analysis indicates that no one was helped. What actually happened is difficult to discern from media statements. For example, the committee stated that their thumbs-down wasn’t due to “any new emergent safety concern, but on a broad assessment of the benefit/risk.” A Roche rep cited an “unfavorable efficacy trend.” I suppose that means that under the conditions of the phase 3 trial, the drug might have lowered huntingtin protein levels, but didn’t have a detectable effect on symptoms or rate of disease progression. I await publication of a paper to learn the details.
Results of the phase 3 study, called GENERATION HD1, were anxiously anticipated because its the largest clinical trial for HD so far. And the findings will be useful no matter what they are. “The data generated will significantly advance our understanding of huntingtin-lowering as a potential treatment approach,” said Levi Garraway, MD, PhD, chief medical officer at Roche. The company has two ongoing trials of the drug at earlier stages.
The second dose of bad news in the HD community came on March 29, when Wave Life Sciences announced that the lowering of the abnormal huntingtin protein in an early phase clinical trial, PRECISION-HD, “didn’t support” further development of two drug candidates. Their technology targets a SNP found in the mutant gene but not the normal version. (A “single nucleotide polymorphism” is one site in a gene that differs in a population.) The treatment is a synthetic small piece of nucleic acid, slightly different from the natural version, that binds to and silences the mRNA transcribed from the mutant gene.
In one trial of 88 participants, the drug didn’t change levels of abnormal huntingtin. In the other trial of 28 people, levels diminished, but “effects were inconsistent.” Analysis of protein level in the cerebrospinal fluid indicated that dosing high enough to affect symptoms safely might not be possible.
These results are disappointing, but further analysis of the data from the discontinued trials could have value. Might analyzing the participants one by one reveal a few who did improve? And if so, what do they share?
Jan Nolta, PhD, director of the stem cell program at the UC Davis School of Medicine and director of the Institute for Regenerative Cures, who works on HD, explains the possible silver lining:
Please stay tuned for further information. Once the data are unblinded, of those receiving the therapy rather than placebo, there might be certain people who were “responders” whereas it wasn’t helping others and that flattened the overall curve – this info needs to be carefully analyzed. The therapy could be truly helping some participants. Then a future trial could be tailored to those fitting those inclusion criteria once it is figured out. It’s important for us all to keep in mind that the trials can be an iterative process.
The seeming setbacks actually mirror how science works: if the hypothesis is disproved, go back to the drawing board and design new experiments to test in other ways.
A microRNA gene therapy strategy
The metaphor of drug development as a pipeline is apt. New options are always arising.
While the HD community has been reeling from the two recent clinical trial disappointments, gene therapy company uniQure announced enrolling the first few patients in a phase 1/2 clinical trial of their treatment for early HD, called AMT-130. It is a gene therapy that is delivered surgically to the striatum, at HD Centers of Excellence, with nine sites in the US and Europe starting in a few months. Patients will be randomized to receive the gene therapy or sham surgery. The blinding – which patient received which intervention – will remain for a year, and then the participants followed for five years, and disease progression compared.
AMT-130 is a viral vector (adeno-associated virus 5) that delivers DNA encoding a microRNA that lowers huntingtin protein. MicroRNAs are tiny RNA molecules that function as “dimmer switches” to control expression (transcription of mRNA) of specific sets of genes. They’re normally in cells, and so AMT-130 harnesses a natural process. The goal is to silence disease-causing genes without harming other genes. The microRNAs may even travel neuron-to-neuron in fleets of exosomes, the tiny natural bubbles that ferry molecules between cells.
To accompany announcement of the start of the clinical trial, a paper in Science Translational Medicine describes the microRNA-based gene therapy in a minipig model. The title is encouraging: “Widespread and sustained target engagement in Huntington’s disease minipigs upon intrastriatal microRNA-based gene therapy.”
MRI revealed that the injected viral vectors were distributed throughout the pigs’ brains and lowered levels of the abnormal huntingtin protein 30% to more than 75%, depending on the brain region. The microRNAs were still dampening protein production a year later. So the clinical trials just getting underway will reveal whether those effects translate into improved symptoms in people.
The halting of clinical trials is devastating news, especially for a rare disease with no treatments. But with eclectic strategies in the works to silence or obliterate the errant expanding gene, researchers will, one day, find an approach that works – or more than one.